应用全基因组DNA互作数据构建染色体三维结构,有助于重现染色质的物理结构,加深对染色质结构的理解,辅助科学研究。
通过DNA-DNA互作数据构建染色体三维结构的方法有很多种,包括MCMC5C、Pastis 、AutoChrom3D、ChromSDE、 ShRec3D 、 ShRec3D+、Chromosome3D、LorDG、Dip-C等等。常见的使用方法还是以Pastis为主。该方法于2014年由Jean-Philippe Vert和William Stafford Noble团队发表,目前其被引用次数已达到259次。

Pastis结合了相互作用的统计模型,通过将相互作用和距离相互转化,可以推断出最能解释观察到的数据的基因组结构。
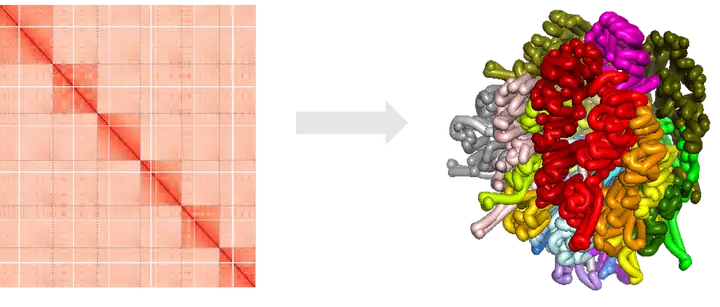
应用Pastis构建基因组3D模型,主要分为三大步骤:将互作矩阵预处理为适用于Pastis分析脚本的输入文件、准备Pastis的配置文件并构建基因组三维结构,最后使用PyMOL进行基因组3D结构的可视化。
1. 互作矩阵预处理
Pastis的输入矩阵仅识别numpy的互作矩阵。对于比对后构建的Hi-C相互作用矩阵,首先需要对其进行预处理。以Juicer流程中用来存储Hi-C互作矩阵信息的二进制文件.hic文件为例:
使用hic2cool将.hic文件转换为cool格式。这里我们保留.hic文件中的多个分辨率,生成包含多个分辨率的.mcool文件。
hic2cool convert ../01.raw_data/4DNFI1UEG1HD.hic 4DNFI1UEG1HD.mcool
对1 Mb分辨率的.cool文件进行Iced balancing。
cooler balance 4DNFI1UEG1HD.mcool::resolutions/1000000
使用cooler api从.cool文件中提取出2D densen Hi-C互作矩阵
python cool2dense_1mb_cooler_iced.py
#cool2dense_1mb_cooler_iced.py脚本中将.coolmatrix转换为numpy 2D matrix;cooler在Iced balancing时进行了rescale,使得contact值变得很小,可以选择不进行rescale或者乘以一个大数用于后续分析。
#脚本获取方式见文末。
得到2D dense文本格式的Hi-C互作矩阵
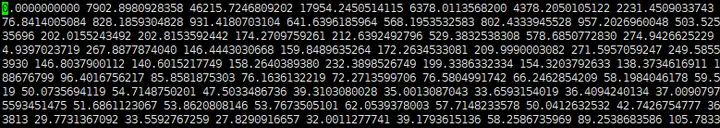
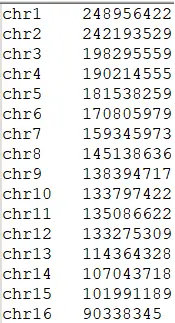
染色体长度文件也是Pastis的输入文件之一。根据分析的物种和参考基因组,准备一个文件,记录每条染色体的长度信息。这个文件可以是一个文本文件,每行是一条染色体的长度,按照染色体编号的顺序排列。
2. Pastis构建基因组三维结构
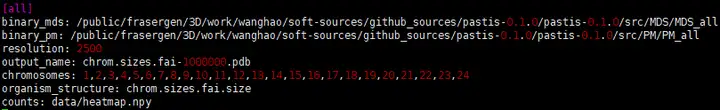
准备好Pastis的输入文件,下一步编写Pastis的配置文件,即在工作目录下,创建一个名为config.ini的配置文件,提供Pastis运行所需的参数。这些参数包括:
• resolution:Hi-C矩阵的分辨率,即每个bin的大小,单位是bp。
• output_name:输出文件的名称,可以是任意字符串。
• chromosomes:要分析的染色体编号,多个用逗号分隔,如1,2,3,4,5,6,7,8。
• organism_structure:染色体长度文件的路径,相对于工作目录或绝对路径。
• counts:矩阵存储为numpy数组格式。
• 此外,还有一些其他可选参数,如nucleus_size(细胞核大小)等。
Pastis提供的四种算法中的任意一种可以用来推断染色体三维结构。这四种算法分别是:
• pastis-mds:基于多维尺度变换(MDS)的算法,将Hi-C矩阵转换为距离矩阵,然后用MDS方法将高维数据降维到三维空间。
• pastis-nmds:基于非度量多维尺度变换(NMDS)的算法,与MDS类似,但不要求距离矩阵满足欧氏空间条件。
• pastis-pm1:基于泊松模型的算法。
• pastis-pm2:基于泊松模型和随机梯度下降的算法。
例如,使用Pastis中的MDS方法来构建基因组3D模型,选择MDS方法依据互作两点间的count进行多重最适化计算,以此来推断出基因组的3D结构。
python 3d.py
#3d.py中根据给定的染色体长度文件和二维矩阵文件2D txt densen,将2D txt densen矩阵转换为pastis输入所需的numpy 2D matrix .npy文件,并生成pastis所需的配置文件和运行脚本,随后运行pastis-MDS后获取可用于可视化的.pdb文件
#脚本获取方式见文末。
Pastis会在工作目录下生成两个输出文件,一个是以npy格式存储的三维坐标数组文件,另一个是以pdb格式存储的三维结构模型文件。后续可以用能够打开pdb格式文件的软件来查看和分析你的三维结构模型,如PyMOL。
3. 基因组三维结构可视化
PyMOL是一个由用户赞助的分子三维结构显示软件。其拥有不同的版本,如商业版、教育版、开源版等,适用于创作高品质的小分子或是生物大分子(特别是蛋白质)的三维结构图像。
PyMOL主页面
PDB文件格式可用于存储蛋白质三维结构,记录了蛋白质中每个原子的坐标信息,蛋白质二级结构信息,以及原子之间的相互作用等信息。
PDB文件的信息记录格式如下:
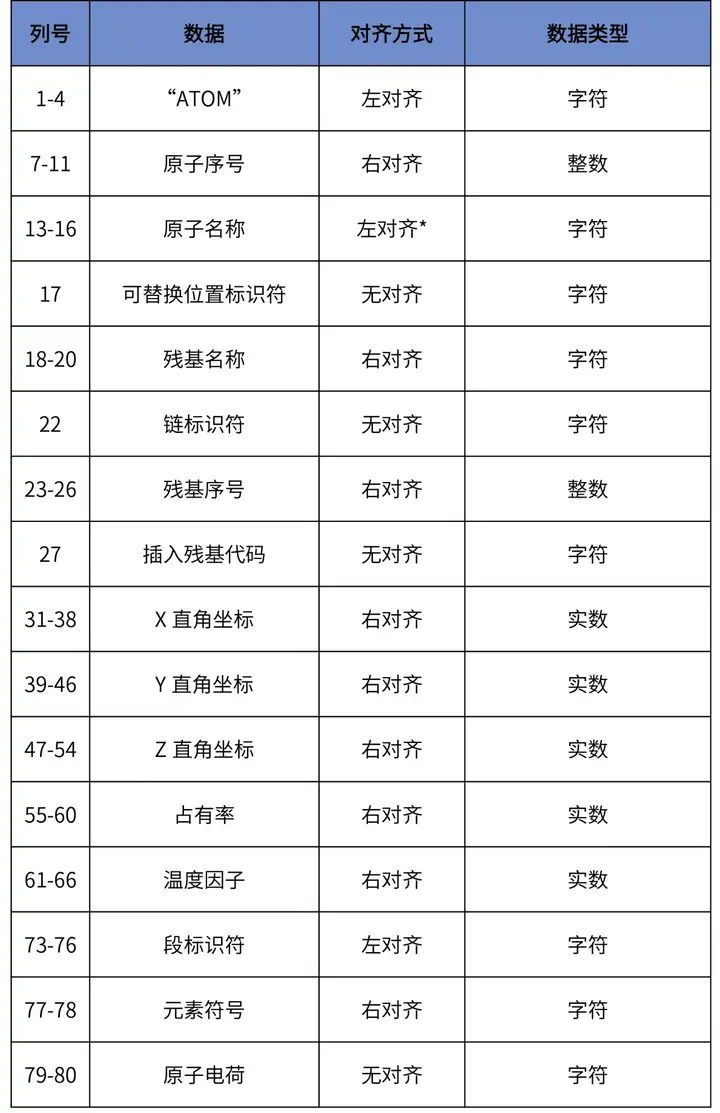
在使用PyMOL对Pastis生成的基因组3D结构.pdb文件进行可视化时,我们主要关注原子序号(Atom serial number,第7-11列)以及链标识符(Chain identifier。第22列)。这两个字段可以帮助我们确定基因组中不同原子和残基的位置和相互作用模式。
PyMOL的主要操作界面分成有两个窗口:控制窗口和显示窗口。控制窗口主要包括菜单栏、操作记录窗口、常用命令面板和命令输入框。显示窗口主要包括可视化窗口、对象列表、模式窗口、动画控制命令窗口。我们可以使用命令行对需要展示的分子进行一系列操作,有关PyMOL相关的命令行操作命令,可以参考:https://pymol.sourceforge.net/newman/user/toc.html。在可视化窗口,通过鼠标操作,我们可以对展示中的分子进行视角变化等调整。
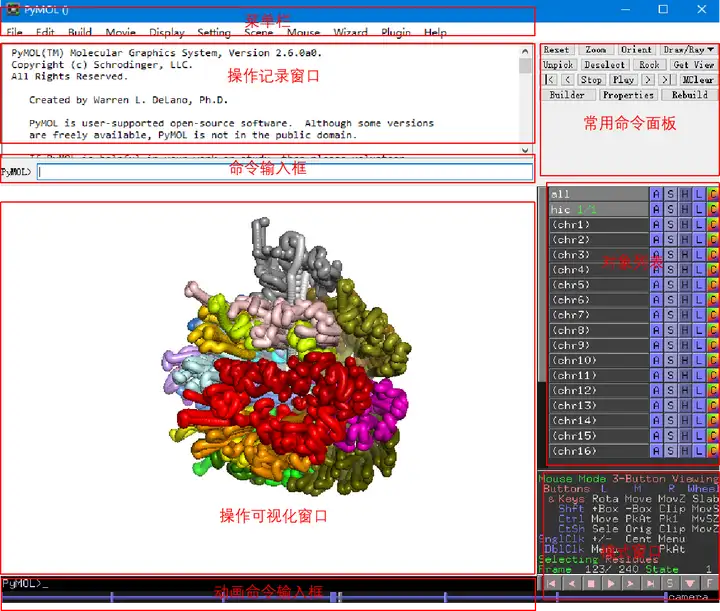
运用Pastis生成的pdb文件在PyMol中进行基因组3D结构模型的可视化。在此过程,通常会需要对染色体进行不同颜色的着色、选取部分染色体进行展示、突出展示可能的目标区域,以及保存结果这四大步骤。
首先,通过PyMOL界面中选取目标.pdb文件,或者直接将.pdb文件拖入到PyMOL窗口中,导入pdb文件。
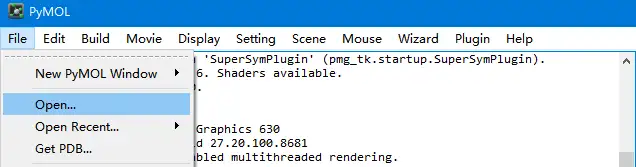
接下来对染色体着色。一方面,我们可以在对象列表点击对象名称来选择,然后点击按钮(C)来进行着色。另一方面,我们可以通过命令行来实现:
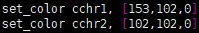
• 设定需要使用的RGB颜色
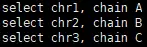
• 选取目标chain,其中其中chr1,chr2等为选定的染色体对象;chain A、B、C等对应pdb文件中的Chain identifier(第22列)
• 进行着色
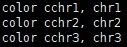
在基因组3D结构可视化中,有时会需要突出显示部分重要区域时,例如着丝粒区域、端粒区域等,这可以通过命令行进行实现,例如高亮显示chr1中的1-200个ATOM,并仅展示chr1,可以按如下步骤:
• 按chain拆分目标分子
split_chains
• 选取目标ATOM
select hilight, chain A & id 1-200
• 进行着色
color hotpink, hilight
• 增大目标区域的sphere大小
alter hilight,vdw=1.8
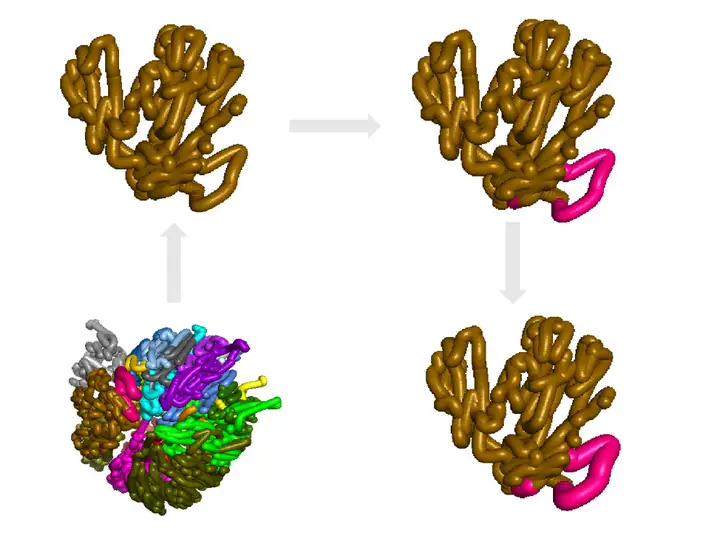
最后,对生成的结果进行保存。
如果想要保存为位图,在选定分子的角度后,在PyMOL的可视化界面点击Draw/Ray设置合适的分辨率,然后选择Draw进行图片的保存操作。
如果想要保存为动态影像,处理好基因组3D模型后,对分子添加movie:
movie.add_roll(4.0,axis=x,start=1)
movie.add_roll(4.0,axis=y,start=121)
#在PyMOL中制作一个沿着x轴旋转的动画的(4.0:表示旋转的时间,单位是秒;axis:表示旋转的轴,可以是x, y, z中的任意一个;start:表示动画开始的帧数。PyMOL默认的帧率是30帧每秒。)
随后在PyMOL可视化界面中选择:File-> Export Movie As -> MPEG,并且应用合适的分辨率进行保存
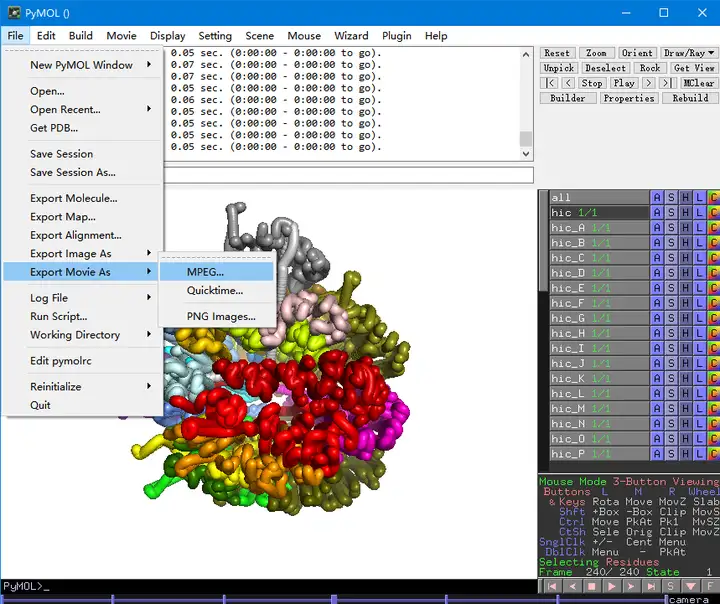
如果想要保存为gif动态图,在上述生成.mpg影响之后,于PyMOL可视化界面中选择:File-> Export Movie As -> PNG Images
在选择好合适的分辨率后保存各个帧的png图片,之后可以将png图片传输到Linux系统,使用Linux下的convert命令将系列png位图转换为.gif动图
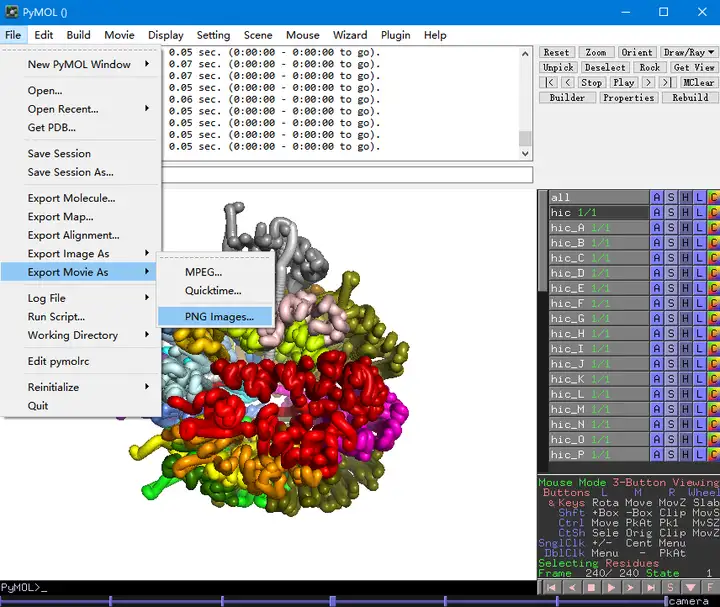
convert pngs/*png hic.gif
总结
以上,我们梳理了应用Pastis构建基因组3D模型并通过PyMOL进行可视化的重点内容。文章描述所涉及的试用数据和详细脚本,在我们今年年中“一元购”的三维基因组公开课中都有分享。感兴趣的老师可通过扫描下方二维码进行获取:
参考
1. https://github.com/hiclib/pastis.
2. http://hiclib.github.io/pastis.
3. Varoquaux N, Ay F, Noble W S, et al. A statistical approach for inferring the 3D structure of the genome[J]. Bioinformatics, 2014, 30(12): i26-i33.
中文官网地址:https://www.ks-vpeptide.com.cn/
英文官网地址:https://www.ks-vpeptide.com
领英:https://www.linkedin.com/company/ks-v-peptide/