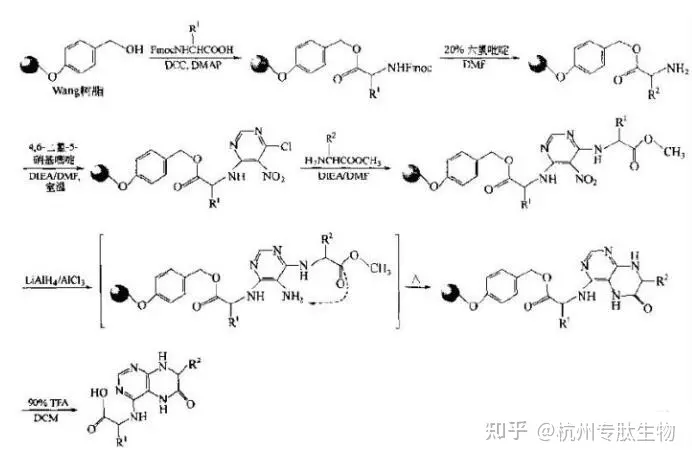
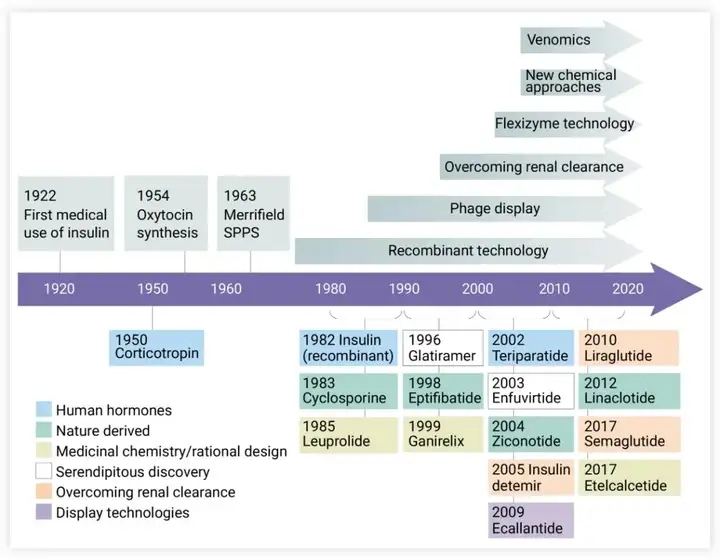
一、简介
现代药物发现包括三个重要步骤:发现与疾病相关的靶点、发现和优化新的先导化合物以及药物开发。基因组学、蛋白组学、分子生物学、细胞生物学、结构生物学、计算生物学和生物信息学方法被用于确定与疾病有关的靶标。此过程中,发病机制的探索和相关靶点的鉴定非常重要。蛋白质错误折叠和聚集是一个动态过程,仅靠实验方法很难进行研究。MD模拟为研究蛋白错误折叠和聚集机制提供了一种高效和方便的方法。MD模拟是研究原子和分子物理运动的有效方法。允许原子和分子在固定的时间内相互作用,从而给出系统动态变化的视图。通常,原子和分子的轨迹是通过数值求解一个相互作用粒子系统的牛顿运动方程来确定的。它们的势能利用不同力场的分子力学方法计算。化学和生物学中最常用的力场是基于分子力学的,且体现了可以再现结构和构象变化的粒子-粒子相互作用的经典力学处理。相对于实验方法,MD模拟不仅可以显示环境变化引起(如pH、温度和残基突变)的结构变化,而且可以显示蛋白质或肽错误折叠和聚集的动态过程。
为了发现和优化新的潜在先导化合物,计算机辅助药物设计已被用于提高效率并减少药物发现的盲目性。一般的分子对接方法缺乏充分考虑靶标的柔性,这可能导致缺少一些活性化合物。相比之下,MD模拟可以获得多个靶标构象,且被广泛用于分子对接以考虑靶标的柔性。能够正确排列候选化合物有效方法的发展一直是虚拟筛选研究的热点。MD模拟结合自由能计算被证明是改善虚拟筛选富集因子的良好策略,因为它可以提高配体与靶标之间结合能力预测的准确性。为了优化先导化合物并获得具有高活性和适宜ADME/T性质的化合物,理解化合物与靶之间的相互作用是非常重要的。MD模拟结合自由能计算已广泛应用于研究药物与靶点之间的相互作用。
二、MD模拟在虚拟筛选中的应用
基于分子对接方法的虚拟筛选已成为药物发现中不可或缺的工具。虚拟筛选的优势在于它可以在很短的时间内处理大规模的化合物库。完整的虚拟筛选过程包括化合物数据库的预处理、分子对接过程以及化合物的选择。每一步都会影响虚拟筛选的性能。一般来说,评估虚拟筛选是否成功很大程度上取决于虚拟筛选的富集因子,富集因子可以量化虚拟筛选方法的有效性。虚拟筛选的每一步都采用了多种策略改善其富集因子。本文主要讨论在分子对接过程的各个步骤中使用MD模拟,以及这种方法如何改进富集因子。常规分子对接期间,根据空间互补性和能量匹配将化
合肥科生景肽生物科技有限公司成立于2018年,目前已经打造了全球领先的以肽为核心的生命分子发现、合成生产、结构优化、递送平台,主要瞄准肽发现及靶向递送,专注于为各大制药企业、生物技术公司、科研单位提供一站式的定制化研发服务。 公司独有的KPDS™平台(KS-V Peptide Discovery Services Platform)是国际领先的的多肽药物发现平台,我们致力于创新药物的高效和精准开发,以科生景肽专有KPDS技术为核心,提供一站式,定制化的多肽发现服务,以灵活的产品形式和服务模式助力广大客户各类药物发现项目的快速推进和应用探究,包括但并不限于疾病诊断及保健功能产品、多肽药物、核素偶联药物(RDC)、基于小分子的肽药物偶联物(PDC)和多功能肽偶联物等。中文官网地址:https://www.ks-vpeptide.com.cn/
英文官网地址:https://www.ks-vpeptide.com
领英:https://www.linkedin.com/company/ks-v-peptide/